Bahan aktif: Esomeprazole
LUCEN 20 mg tablet tahan gastro
Tablet tahan gastro LUCEN 40 mg
Sisipan pakej Lucen tersedia untuk saiz pek: - LUCEN 20 mg tablet tahan gastro, LUCEN 40 mg tablet tahan gastro
- LUCEN 10 mg butiran tahan gastro untuk penggantungan oral, dalam sachet
- LUCEN 40 mg serbuk untuk penyelesaian untuk suntikan / infus
Petunjuk Mengapa Lucen digunakan? Untuk apa itu?
LUCEN mengandungi ubat yang disebut esomeprazole. Ia tergolong dalam kumpulan ubat yang disebut 'proton pump inhibitor', yang berfungsi dengan mengurangkan jumlah asid yang dihasilkan oleh perut.
LUCEN digunakan untuk rawatan gangguan berikut:
- "Penyakit refluks gastroesophageal" (GERD). Ia berlaku apabila asid dari perut keluar ke kerongkongan (tiub yang menghubungkan tekak ke perut), menyebabkan rasa sakit, radang dan terbakar.
- Ulser perut atau usus besar yang dijangkiti bakteria yang disebut "Helicobacter pylori". Sekiranya anda mempunyai keadaan ini, doktor anda juga boleh menetapkan antibiotik untuk merawat jangkitan dan membiarkan ulser sembuh.
- Ulser perut disebabkan oleh ubat-ubatan yang disebut NSAID (Non-Steroidal Anti-Inflammatory Drugs).LUCEN juga boleh digunakan untuk mencegah ulser perut terbentuk semasa mengambil NSAID.
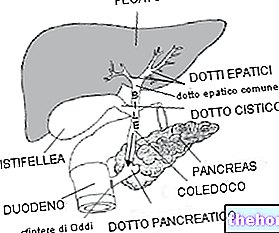
- Asid perut yang berlebihan disebabkan oleh tumor di pankreas (sindrom Zollinger-Ellisson).
- Rawatan pendarahan ulser yang berpanjangan, selepas pencegahan dengan pemberian Lucen secara intravena
Kontraindikasi Apabila Lucen tidak boleh digunakan
Jangan mengambil LUCEN:
- jika anda alah (hipersensitif) terhadap esomeprazole atau mana-mana ramuan lain dari ubat ini (disenaraikan dalam bahagian: Maklumat lanjut).
- jika anda alah kepada ubat penghambat pam proton lain (mis. pantoprazole, lansoprazole, rabeprazole, omeprazole).
- jika anda mengambil ubat yang mengandungi nelfinavir (digunakan untuk merawat HIV).
Anda tidak boleh mengambil LUCEN jika berlaku dalam salah satu kes di atas. Sekiranya ragu-ragu, berjumpa dengan doktor atau ahli farmasi anda sebelum mengambil LUCEN.
Langkah berjaga-jaga untuk penggunaan Apa yang perlu anda ketahui sebelum mengambil Lucen
Berhati-hati dengan LUCEN
Bercakap dengan doktor atau ahli farmasi anda sebelum mengambil LUCEN jika:
- Anda mempunyai masalah hati yang teruk
- Anda mengalami masalah buah pinggang yang teruk.
LUCEN dapat menyembunyikan gejala penyakit lain. Oleh itu, jika ada perkara berikut berlaku kepada anda sebelum anda mula mengambil atau semasa anda mengambil LUCEN, beritahu doktor anda dengan segera:
- Anda menurunkan banyak berat badan tanpa sebab atau menghadapi masalah menelan
- Sakit perut atau senak berlaku
- Mula muntah makanan atau darah
- Najis berwarna hitam (najis berlumuran darah).
Sekiranya anda telah diresepkan LUCEN secara "mengikut keperluan", hubungi doktor anda sekiranya gejala berlanjutan atau mengubah ciri.
Sekiranya anda mengambil perencat pam proton seperti LUCEN, terutamanya selama lebih dari satu tahun, anda mungkin mempunyai sedikit risiko patah tulang pinggul, pergelangan tangan atau tulang belakang. Sekiranya anda menghidap osteoporosis atau mengambil kortikosteroid (yang boleh meningkatkan risiko osteoporosis) berjumpa doktor
Interaksi Dadah atau makanan mana yang boleh mengubah kesan Lucen
Beritahu doktor atau ahli farmasi anda jika anda mengambil atau baru-baru ini mengambil ubat lain, termasuk ubat yang diperoleh tanpa preskripsi.
Sesungguhnya, LUCEN boleh mempengaruhi cara beberapa ubat berfungsi dan beberapa ubat boleh memberi kesan kepada LUCEN. Anda tidak boleh mengambil LUCEN jika anda mengambil ubat yang mengandungi nelfinavir (digunakan untuk merawat HIV).
Beritahu doktor atau ahli farmasi anda jika anda mengambil ubat berikut:
- Atazanavir (digunakan untuk merawat HIV)
- Clopidogrel (digunakan untuk mencegah pembekuan darah)
- Ketoconazole, itraconazole atau voriconazole (digunakan untuk merawat jangkitan yang disebabkan oleh kulat).
- Erlotinib (digunakan untuk merawat barah).
- Citalopram, imipramine atau clomipramine (digunakan untuk merawat kemurungan).
- Diazepam (digunakan untuk rawatan kegelisahan, untuk relaksasi otot atau dalam epilepsi).
- Phenytoin (digunakan dalam epilepsi) Sekiranya anda mengambil fenitoin, doktor anda perlu memantau anda ketika memulakan atau menghentikan rawatan dengan LUCEN.
- Ubat yang digunakan untuk mengencerkan darah, seperti warfarin. Doktor anda mungkin memantau anda semasa anda memulakan atau menghentikan rawatan dengan LUCEN.
- Cilostazol (digunakan untuk merawat claudication sekejap - sakit di kaki ketika berjalan kerana bekalan darah tidak mencukupi).
- Cisapride (digunakan untuk pencernaan dan pedih ulu hati).
- Digoxin (digunakan untuk masalah jantung).
- Methotrexate (ubat kemoterapi yang digunakan dalam dos tinggi untuk merawat barah) - jika anda mengambil metotreksat dalam dos yang tinggi, doktor anda boleh menghentikan rawatan anda dengan Lucen buat sementara waktu.
- Tacrolimus (digunakan dalam pemindahan organ)
- Rifampicin (digunakan untuk merawat batuk kering).
- St John's wort (Hypericum perforatum) (digunakan untuk merawat kemurungan).
Sekiranya doktor anda telah menetapkan antibiotik seperti amoksisilin dan klaritromisin dengan LUCEN untuk rawatan bisul yang disebabkan oleh jangkitan Helicobacter pylori, sangat penting anda memberitahu doktor mengenai ubat lain.
Amaran Penting untuk mengetahui bahawa:
Kehamilan dan penyusuan
Sebelum mengambil LUCEN, beritahu doktor anda jika anda hamil atau ingin hamil. Minta nasihat doktor atau ahli farmasi anda sebelum mengambil sebarang ubat. Doktor anda akan memutuskan sama ada anda boleh mengambil LUCEN selama ini.
Tidak diketahui sama ada LUCEN masuk ke dalam susu ibu, oleh itu anda tidak boleh mengambil LUCEN jika anda menyusu.
Mengambil LUCEN dengan makanan dan minuman
Tablet boleh diambil semasa perut penuh atau semasa perut kosong.
Memandu dan menggunakan mesin
LUCEN tidak mungkin mempengaruhi kemampuan anda untuk memandu atau menggunakan alat atau mesin apa pun.
Maklumat penting mengenai beberapa ramuan LUCEN
Tablet tahan gastrik LUCEN mengandungi sukrosa yang merupakan sejenis gula. Sekiranya anda diberitahu oleh doktor bahawa anda mempunyai "intoleransi terhadap beberapa gula, berjumpa dengannya sebelum mengambil ubat.
Dos dan kaedah penggunaan Cara penggunaan Lucen: Dos
Sentiasa mengambil LUCEN tepat seperti yang diberitahu oleh doktor anda. Sekiranya ragu-ragu, anda harus berjumpa doktor atau ahli farmasi anda.
- Tablet tahan gastrik LUCEN tidak digalakkan untuk kanak-kanak di bawah 12 tahun
- Sekiranya anda telah lama mengambil ubat ini, doktor anda akan memantau anda (terutamanya jika anda telah mengambil ubat ini lebih dari satu tahun)
- Sekiranya doktor anda memberitahu anda untuk mengambil ubat apabila diperlukan, jika perlu, beritahu doktor anda jika gejala anda berubah.
Mengambil ubat
- Anda boleh mengambil tablet pada bila-bila masa sepanjang hari.
- Anda boleh mengambil tablet semasa perut penuh atau semasa perut kosong.
- Telan keseluruhan tablet dengan minum air. Jangan mengunyah atau menghancurkan tablet kerana ia mengandungi butiran bersalut yang melindungi ubat daripada keasidan gastrik. Oleh itu, penting untuk tidak merosakkan butiran.
Apa yang perlu dilakukan sekiranya anda menghadapi masalah menelan tablet
Sekiranya anda menghadapi masalah menelan tablet:
- Masukkan tablet ke dalam segelas air pegun. Cecair lain tidak boleh digunakan
- Kacau sehingga tablet larut (campuran tidak akan kelihatan jelas). Minum segera atau sekurang-kurangnya dalam masa 30 minit. Sentiasa campurkan mereka sebelum minum
- Untuk memastikan bahawa anda telah mengambil semua ubat, bilas gelas dengan bersih dengan mengisinya separuh dengan air dan minum. Zarah pepejal mengandungi ubat dan tidak boleh dikunyah atau dihancurkan.
Sekiranya anda benar-benar tidak dapat menelan, tablet boleh dicampurkan dengan sedikit air, dimasukkan ke dalam picagari dan diberikan melalui tiub terus ke perut (saluran gastrik).
Berapa banyak ubat yang perlu diambil
- Doktor anda akan memberitahu anda mengenai jumlah tablet yang akan diambil dan berapa lama. Ini adalah fungsi dari keadaan fizikal, usia dan keadaan hati anda.
- Dos biasa diberikan di bawah.
Rawatan pedih ulu hati yang disebabkan oleh penyakit gastroesophageal reflux (GERD):
Dewasa dan kanak-kanak dari 12 tahun:
- Sekiranya doktor anda mendapati kerongkongan anda sedikit rosak, dos biasa adalah satu tablet tahan gastrik LUCEN 40 mg sekali sehari selama 4 minggu. Doktor anda mungkin memberitahu anda untuk meneruskan rawatan, dengan dos yang sama, selama 4 minggu lagi, sekiranya esofagus tidak sembuh.
- Selepas penyembuhan kerongkongan, dos yang biasa adalah satu tablet tahan gastro LUCEN 20 mg sekali sehari.
- Sekiranya kerongkongan tidak rosak, dos biasa adalah satu tablet tahan gastrik LUCEN 20 mg setiap hari. Apabila gejala terkawal, doktor anda akan memberitahu anda bahawa anda boleh minum ubat tersebut apabila diperlukan, sehingga maksimum satu gastro- tablet tahan Lucen 20 mg sehari.
- Sekiranya anda mempunyai masalah hati yang teruk, doktor anda akan memberi anda dos yang lebih rendah.
Rawatan bisul yang disebabkan oleh jangkitan Helicobacter pylori dan pencegahan kemunculan semula:
- Orang dewasa dari usia 18 tahun dan seterusnya: dos biasa adalah satu tablet tahan gastrik LUCEN 20 mg dua kali sehari selama satu minggu.
- Doktor anda juga akan memberitahu anda untuk mengambil antibiotik yang disebut amoxicillin dan clarithromycin.
Rawatan ulser gastrik yang disebabkan oleh NSAID (Dadah Anti-Radang Bukan Steroid):
- Orang dewasa dari usia 18 tahun dan seterusnya: dos biasa adalah satu tablet tahan gastrik LUCEN 20 mg sekali sehari selama 4 hingga 8 minggu.
Pencegahan ulser perut jika anda mengambil NSAID (Dadah Anti-Radang Bukan Steroid):
- Dewasa dari usia 18 tahun dan seterusnya: dos biasa adalah satu tablet tahan gastrik LUCEN 20 mg sekali sehari.
Rawatan lebihan asid perut yang disebabkan oleh pertumbuhan pankreas (sindrom Zollinger-Ellison):
- Orang dewasa dari usia 18 tahun dan seterusnya: dos biasa adalah satu tablet tahan gastrik LUCEN 40 mg dua kali sehari.
- Doktor anda akan menyesuaikan dos mengikut keperluan anda dan juga akan memutuskan berapa lama untuk meneruskan rawatan.
Dos maksimum adalah 80 mg dua kali sehari.
Rawatan pendarahan ulser yang berpanjangan, selepas pencegahan dengan pemberian Lucen secara intravena:
Dos biasa adalah satu tablet Lucen 40 mg sekali sehari selama 4 minggu.
Overdosis Apa yang perlu dilakukan sekiranya anda mengambil terlalu banyak Lucen
Sekiranya anda mengambil LUCEN lebih banyak dari yang sepatutnya
Sekiranya anda telah mengambil lebih banyak LUCEN daripada yang ditetapkan oleh doktor anda, beritahu doktor atau ahli farmasi anda dengan segera.
Sekiranya anda terlupa mengambil LUCEN
- Sekiranya anda terlupa mengambil dos LUCEN, ambil secepat yang anda ingat. Sekiranya sudah hampir masanya untuk dos seterusnya, lewati dos yang tidak dijawab.
- Jangan mengambil dos berganda (dua dos secara serentak) untuk menebus dos yang dilupakan.
Kesan Sampingan Apakah kesan sampingan Lucen
Seperti semua ubat, LUCEN boleh menyebabkan kesan sampingan, walaupun tidak semua orang mendapatnya.
Sekiranya anda melihat kesan sampingan serius berikut, hentikan pengambilan LUCEN dan segera hubungi doktor anda:
- Mengi tiba-tiba, bengkak bibir, lidah dan tekak atau badan, ruam, pingsan atau sukar menelan (reaksi alahan teruk).
- Kemerahan kulit dengan lepuh atau mengelupas. Lepuh dan pendarahan yang teruk juga boleh muncul di bibir, mata, mulut, hidung dan alat kelamin. Ini mungkin "sindrom Stevens-Johnson" atau "nekrolisis epidermis toksik".
- Kulit kuning, urin gelap, dan keletihan boleh menjadi gejala masalah hati. Kesan ini jarang berlaku, mempengaruhi kurang dari 1 dalam 1000 orang.
Kesan sampingan lain termasuk:
Biasa (mempengaruhi kurang dari 1 dalam 10 orang):
- Sakit kepala.
- Kesan pada perut atau usus: cirit-birit, sakit perut, sembelit, perut kembung.
- Mual atau muntah.
Tidak biasa (mempengaruhi kurang daripada 1 dari 100 orang):
- Bengkak di kaki dan pergelangan kaki.
- Tidur terganggu (insomnia).
- Pening, pin dan jarum, mengantuk.
- Pening.
- Mulut kering
- Perubahan dalam ujian darah yang memeriksa bagaimana hati berfungsi.
- Ruam kulit, gatal-gatal dan gatal-gatal.
- Patah tulang pinggul, pergelangan tangan atau tulang belakang (jika Lucen digunakan dalam dos yang tinggi dan untuk jangka masa yang lama).
Jarang (mempengaruhi kurang dari 1 dalam 1,000 orang):
- Masalah darah, seperti penurunan jumlah sel darah putih dan platelet. Ini boleh menyebabkan kelemahan, lebam, atau kemungkinan mendapat jangkitan dengan lebih mudah.
- Tahap natrium rendah dalam darah. Ini boleh menyebabkan kelemahan, muntah dan kekejangan.
- Rasa terkilan, keliru atau tertekan.
- Perubahan rasa.
- Masalah dengan penglihatan anda, seperti penglihatan kabur.
- Mengi tiba-tiba atau sesak nafas (bronkospasme).
- Keradangan bahagian dalam mulut.
- Jangkitan yang disebut "thrush" yang boleh mempengaruhi usus dan disebabkan oleh jamur.
- Masalah hati, termasuk penyakit kuning yang boleh menyebabkan kulit kuning, kencing gelap dan keletihan.
- Kehilangan rambut (alopecia).
- Ruam kulit pada pendedahan cahaya matahari.
- Sakit sendi (arthralgia) atau sakit otot (myalgia).
- Perasaan umum tidak sihat dan kekurangan kekuatan.
- Berpeluh bertambah.
Sangat jarang berlaku (mempengaruhi kurang dari 1 dari 10,000 orang):
- Perubahan jumlah sel darah, termasuk agranulositosis (kekurangan sel darah putih).
- Pencerobohan.
- Melihat, merasakan atau mendengar perkara yang tidak ada (halusinasi).
- Masalah hati yang teruk menyebabkan kegagalan hati dan keradangan otak.
- Muncul secara tiba-tiba ruam yang teruk atau melepuh atau mengelupas kulit. Ini mungkin berkaitan dengan demam tinggi dan sakit sendi (eritema multiforme, sindrom Stevens-Johnson, nekrolisis epidermis toksik).
- Kelemahan otot.
- Masalah buah pinggang yang teruk.
- Pembesaran payudara pada lelaki.
Tidak diketahui (frekuensi tidak dapat dianggarkan dari data yang ada)
- Sekiranya anda mengambil LUCEN selama lebih dari tiga bulan, kadar magnesium dalam darah anda mungkin menurun. Tahap magnesium yang rendah dapat menampakkan diri dengan keletihan, kontraksi otot yang tidak disengajakan, disorientasi, kejang, pening, peningkatan kadar jantung. Sekiranya anda mempunyai simptom-simptom ini, segera berjumpa doktor. Tahap magnesium yang rendah juga boleh menyebabkan penurunan kadar kalium atau kalsium dalam darah. Doktor anda harus memutuskan sama ada memeriksa kadar magnesium darah anda secara berkala.
- Keradangan pada usus (membawa kepada cirit-birit).
LUCEN dalam kes yang sangat jarang mempengaruhi sel darah putih yang menyebabkan kekurangan imuniti. Sekiranya anda mengalami jangkitan dengan gejala seperti demam dengan kemerosotan keadaan fizikal umum anda atau demam dengan gejala jangkitan tempatan, seperti sakit di leher, tekak atau mulut atau kesukaran membuang air kecil, anda harus berjumpa doktor secepat mungkin sehingga bahawa kekurangan sel darah putih (agranulositosis) dapat dikesampingkan melalui ujian darah. Penting bagi anda untuk memberikan maklumat mengenai ubat-ubatan yang anda ambil. Jangan risau tentang senarai kesan sampingan di atas. Kemungkinan tidak akan muncul.Sekiranya ada kesan sampingan yang serius, atau jika anda melihat kesan sampingan yang tidak disenaraikan dalam risalah ini, sila beritahu doktor atau ahli farmasi anda.
Tamat Tempoh dan Pengekalan
- Jauhkan dari jarak dan jangkauan kanak-kanak.
- Jangan simpan di atas 30 ° C.
- Simpan dalam bungkusan asal (lepuh) atau simpan bekas dengan rapat (botol) untuk melindungi dari kelembapan.
- Jangan gunakan tablet selepas tarikh luput (EXP) yang tertera pada kadbod, dompet atau lepuh. Tarikh luput merujuk pada hari terakhir dalam sebulan.
- Ubat tidak boleh dibuang melalui air sisa atau sampah isi rumah. Tanya ahli farmasi anda bagaimana membuang ubat yang tidak lagi anda gunakan. Ini akan membantu melindungi alam sekitar.
MAKLUMAT LAIN
Apa yang mengandungi LUCEN
Bahan aktif adalah esomeprazole. Tablet tahan gastrik LUCEN terdapat dalam 2 kekuatan yang mengandungi 20 atau 40 mg esomeprazole (sebagai magnesium trihydrate).
Bahan-bahan lain adalah: gliserol monostearat 40-55, hyprolose, hypromellose, iron oxide (red-brown, yellow) (E172, only for 20 mg tablet), magnesium stearate, methacrylic acid copolymer ethyl acrylate (1: 1) dispersi pada 30 %, selulosa mikrokristal, parafin sintetik, makrogol, polysorbate 80, crospovidone, sodium stearyl fumarate, scrose sferes (sukrosa dan jagung kanji), talc, titanium dioksida (E171), triethyl citrate.
Penerangan mengenai penampilan LUCEN dan kandungan peknya
- Tablet tahan gastro LUCEN 20 mg berwarna merah muda muda dengan A / EH di satu sisi dan 20 mg di sisi lain.
- Tablet tahan gastro LUCEN 40 mg berwarna merah jambu dengan A / EI di satu sisi dan 40 mg di sisi lain.
- Tablet terdapat dalam pek lepuh, dompet dan / atau botol yang berisi
- 20 mg, 40 mg: sebotol tablet 2-5-7-14-15-28-30-56-60-100-140 (28x5).
- 20 mg, 40 mg: lepuh atau lepuh dompet 3-7-7x1-14-15-25x1-28-30-50x1- 56-60-90-98-100x1-140 tablet.
Tidak semua saiz pek boleh dipasarkan
Risalah Pakej Sumber: AIFA (Badan Perubatan Itali). Kandungan yang diterbitkan pada Januari 2016. Maklumat yang ada mungkin tidak terkini.
Untuk mempunyai akses ke versi paling terkini, disarankan untuk mengakses laman web AIFA (Badan Perubatan Itali). Penafian dan maklumat berguna.
01.0 NAMA PRODUK PERUBATAN
LUCENTIS 10 MG / ML PENYELESAIAN UNTUK INJEKSI
02.0 KOMPOSISI KUALITATIF DAN KUANTITATIF
Satu ml mengandungi 10 mg ranibizumab *. Setiap botol mengandungi 2.3 mg ranibizumab dalam 0.23 ml larutan.
* Ranibizumab adalah fragmen antibodi monoklonal manusiawi yang dihasilkan dalam sel Escherichia coli oleh teknologi DNA rekombinan.
Untuk senarai lengkap eksipien, lihat bahagian 6.1.
03.0 BORANG FARMASI
Penyelesaian suntikan
Larutan akueus jernih, tidak berwarna hingga kuning pucat.
04.0 MAKLUMAT KLINIKAL
04.1 Petunjuk terapeutik
Lucentis ditunjukkan pada orang dewasa untuk:
• Rawatan degenerasi makula neovaskular (basah) yang berkaitan dengan usia
• Rawatan gangguan penglihatan yang disebabkan oleh edema makula diabetes (DME)
• Rawatan gangguan penglihatan yang disebabkan oleh "edema makula sekunder kepada oklusi vena retina (RVO cabang atau RVO tengah)
• Rawatan gangguan penglihatan yang disebabkan oleh neovaskularisasi choroidal (CNV) yang disebabkan oleh miopia patologi (PM)
04.2 Posologi dan kaedah pentadbiran
Lucentis harus diberikan oleh pakar oftalmologi berkelayakan yang berpengalaman dalam suntikan intravitreal.
Posologi untuk rawatan AMD basah
Dos Lucentis yang disyorkan ialah 0.5 mg diberikan setiap bulan sebagai suntikan intravitreal tunggal. Ini sesuai dengan isipadu yang disuntikkan sebanyak 0,05 ml.
Rawatan diberikan setiap bulan dan dilanjutkan sehingga ketajaman penglihatan maksimum dicapai iaitu ketajaman penglihatan pesakit stabil selama tiga bulan pemeriksaan berturut-turut yang dilakukan semasa rawatan ranibizumab.
Oleh itu, ketajaman penglihatan pesakit harus dipantau setiap bulan.
Rawatan harus dilanjutkan apabila pemantauan menunjukkan penurunan ketajaman penglihatan akibat AMD basah. Suntikan bulanan kemudiannya harus diberikan sehingga ketajaman visual yang stabil dicapai sekali lagi untuk tiga kali pemeriksaan bulanan berturut-turut (ini menunjukkan minimum dua suntikan). Selang antara dua dos tidak boleh kurang dari satu bulan.
Posologi untuk rawatan gangguan penglihatan yang disebabkan oleh DME atau edema makula sekunder RVO
Dos Lucentis yang disyorkan ialah 0.5 mg diberikan setiap bulan sebagai suntikan intravitreal tunggal. Ini sesuai dengan isipadu yang disuntikkan sebanyak 0,05 ml.
Rawatan diberikan setiap bulan dan dilanjutkan sehingga ketajaman penglihatan maksimum dicapai iaitu ketajaman penglihatan pesakit stabil selama tiga bulan pemeriksaan berturut-turut yang dilakukan semasa rawatan ranibizumab. Sekiranya tidak ada peningkatan ketajaman penglihatan selama tiga suntikan pertama, rawatan tidak digalakkan.
Oleh itu, ketajaman penglihatan pesakit harus dipantau setiap bulan.
Rawatan harus dilanjutkan apabila pemantauan menunjukkan penurunan ketajaman penglihatan disebabkan oleh DME atau edema makular sekunder daripada RVO. Suntikan bulanan kemudian harus diberikan sehingga ketajaman visual yang stabil dicapai sekali lagi untuk tiga pemeriksaan bulanan. Berturut-turut (ini melibatkan minimum dua suntikan). Selang antara dua dos tidak boleh kurang dari satu bulan.
Lucentis dan laser photocoagulation di DME dan edema makula sekunder BRVO
Terdapat beberapa pengalaman pemberian Lucentis bersamaan dengan photocoagulation laser (lihat bahagian 5.1). Apabila diberikan pada hari yang sama, Lucentis harus diberikan sekurang-kurangnya 30 minit selepas photocoagulation laser. Lucentis boleh diberikan kepada pesakit yang sebelumnya telah menerima laser photocoagulation.
Posologi untuk rawatan gangguan penglihatan yang disebabkan oleh CNV sekunder hingga PM
Rawatan harus dimulakan dengan satu suntikan.
Sekiranya pemantauan menunjukkan tanda-tanda aktiviti penyakit, seperti penurunan ketajaman penglihatan dan / atau tanda-tanda kecederaan, rawatan lebih lanjut dianjurkan.
Pemantauan penyakit boleh merangkumi pemeriksaan klinikal, tomografi koheren optik (OCT), atau angiografi fluorescein (FA).
Walaupun sesetengah pesakit hanya memerlukan satu atau dua suntikan pada tahun pertama rawatan, ada yang memerlukan rawatan yang lebih kerap (lihat bahagian 5.1). Oleh itu, pemantauan bulanan disarankan untuk dua bulan pertama dan sekurang-kurangnya setiap tiga bulan pada tahun pertama rawatan. Selepas tahun pertama, kekerapan pemantauan dapat ditentukan oleh doktor.
Selang antara dua dos tidak boleh kurang dari satu bulan.
Lucentis dan terapi fotodinamik dengan Visudyne di CNV sekunder hingga PM
Tidak ada pengalaman dengan pentadbiran Lucentis dalam kombinasi dengan Visudyne.
Populasi khas
Kekurangan hepatik
Lucentis belum dikaji pada pesakit dengan kekurangan hepatik. Walau bagaimanapun, tidak perlu pertimbangan khas untuk penggubalan ini.
Kegagalan buah pinggang
Tidak perlu penyesuaian dos pada pesakit dengan kekurangan buah pinggang (lihat bahagian 5.2).
Warga emas
Tidak perlu penyesuaian dos pada orang tua. Terdapat pengalaman "terhad" pada pesakit dengan DME yang berusia lebih dari 75 tahun.
Populasi kanak-kanak
Keselamatan dan keberkesanan Lucentis pada kanak-kanak dan remaja di bawah 18 tahun belum dapat dipastikan. Tidak ada data yang tersedia.
Kaedah pentadbiran
Botol penggunaan tunggal untuk penggunaan intravitreal sahaja.
Sebelum pentadbiran Lucentis harus diperiksa secara visual untuk mengetahui adanya zarah dan perubahan warna.
Prosedur suntikan mesti dilakukan dalam keadaan aseptik, termasuk pembasmian kuman tangan seperti prosedur pembedahan, sarung tangan steril, tirai steril dan blepharostat steril (atau setara) dan kemungkinan melakukan paracentesis steril (jika perlu riwayat reaksi hipersensitiviti pesakit harus dinilai dengan teliti sebelum prosedur intravitreal (lihat bahagian 4.4). Anestesia yang mencukupi dan antimikroba topikal spektrum luas harus diberikan sebelum suntikan untuk membasmi kuman pada permukaan periokular, okular dan kelopak mata, seperti dalam praktik klinikal.
Untuk maklumat mengenai penyediaan Lucentis, lihat bahagian 6.6.
Masukkan jarum suntikan 3,5-4,0 mm posterior ke limbus, ke dalam ruang vitreous, mengelakkan meridian mendatar dan mengarahkan jarum ke arah pusat bola mata. Suntikan jumlah suntikan 0,05 ml; ubah tempat skleral untuk suntikan berikutnya.
04.3 Kontraindikasi
Hipersensitiviti terhadap bahan aktif atau salah satu daripada eksipien yang disenaraikan dalam bahagian 6.1.
Pesakit dengan jangkitan okular atau periokular semasa atau disyaki.
Pesakit dengan keradangan intraokular yang teruk.
04.4 Amaran khas dan langkah berjaga-jaga yang sesuai untuk digunakan
Tindak balas yang berkaitan dengan suntikan intravitreal
Suntikan intravitreal, termasuk dengan Lucentis, telah dikaitkan dengan endophthalmitis, keradangan intraokular, detasmen retina rhegmatogenous, pecah retina dan katarak traumatik iatrogenik (lihat bahagian 4.8). Teknik suntikan aseptik yang sesuai harus selalu digunakan untuk pentadbiran Lucentis. Sebagai tambahan, pesakit harus dipantau pada minggu setelah suntikan untuk membolehkan rawatan yang cepat sekiranya berlaku jangkitan. Pesakit harus diberi petunjuk tentang cara melaporkan sebarang gejala yang menunjukkan endophthalmitis atau kejadian di atas tanpa berlengah.
Peningkatan tekanan intraokular
Peningkatan sementara tekanan intraokular (IOP) telah diperhatikan dalam 60 minit selepas suntikan Lucentis. Peningkatan IOP yang berpanjangan juga telah diperhatikan (lihat bahagian 4.8). Tekanan intraokular dan perfusi kepala saraf optik harus dipantau dan dirawat dengan tepat.
Rawatan dua hala
Data terhad mengenai penggunaan Lucentis dua hala (termasuk dos pada hari yang sama) tidak menunjukkan peningkatan risiko kejadian buruk sistemik berbanding dengan rawatan unilateral.
Imunogenik
Terdapat potensi imunogenik dengan Lucentis. Oleh kerana terdapat kemungkinan peningkatan pendedahan sistemik pada subjek dengan DME, peningkatan risiko terkena hipersensitiviti pada populasi pesakit ini tidak dapat dikecualikan.Pasien juga harus dididik tentang cara melaporkan jika keradangan intraokular bertambah buruk kerana ia mungkin merupakan gejala klinikal yang disebabkan pembentukan antibodi intraokular.
Penggunaan bersamaan dengan anti-VEGF lain (faktor pertumbuhan endotel vaskular)
Lucentis tidak boleh diberikan bersamaan dengan produk ubat anti-VEGF lain (sistemik atau okular).
Penamatan Lucentis
Dos tidak boleh diberikan dan rawatan tidak boleh dilanjutkan sebelum rawatan berjadual berikutnya sekiranya:
• penurunan ketajaman visual (BCVA) correct30 yang diperbetulkan dengan terbaik berbanding dengan penilaian terakhir;
• tekanan intraokular ≥30 mmHg;
• rehat retina;
• "pendarahan subretinal meluas ke pusat fovea, atau jika tahap pendarahan adalah ≥50% dari keseluruhan kawasan lesi";
• pembedahan intraokular yang dilakukan atau dirancang dalam 28 hari sebelumnya atau berikutnya.
Pecahnya epitel pigmen retina
Faktor risiko yang berkaitan dengan permulaan pecahnya epitel pigmen retina berikutan terapi anti-VEGF untuk AMD basah termasuklah detasmen epitel pigmen retina yang besar dan / atau tinggi. Semasa memulakan terapi dengan Lucentis, berhati-hati harus digunakan pada pesakit dengan faktor risiko ini untuk pecahnya epitel pigmen retina.
Detasmen retina regmatogen atau lubang makula
Rawatan harus dihentikan pada individu dengan detasmen retina rhegmatogenous atau lubang makula tahap 3 atau 4.
Populasi dengan data terhad
Hanya terdapat pengalaman terhad dalam rawatan subjek dengan DME sekunder untuk diabetes jenis I. Lucentis belum pernah dikaji pada pesakit yang sebelumnya telah menerima suntikan intravitreal, pada pesakit dengan jangkitan sistemik aktif, retinopati diabetes proliferatif, atau pada pesakit dengan keadaan perubatan yang bersamaan seperti detasmen retina atau lubang makula.Tiada juga pengalaman dalam rawatan dengan Lucentis pada pesakit diabetes dengan HbAlc lebih besar daripada 12% dan hipertensi tidak terkawal. Kekurangan maklumat harus dipertimbangkan oleh doktor semasa merawat pesakit ini.
Pada pesakit PM, terdapat data terhad mengenai kesan Lucentis pada pesakit yang sebelumnya dirawat dengan terapi fotodinamik yang tidak berjaya dengan verteporfin (vPDT). Selanjutnya, sementara kesan yang konsisten diperhatikan pada subjek dengan lesi subfoveal dan juxtafoveal, data tidak mencukupi mengenai kesannya Lucentis dalam subjek PM dengan lesi extrafoveal.
Kesan sistemik berikutan pentadbiran intravitreal
Kejadian buruk sistemik termasuk pendarahan bukan okular dan kejadian tromboemboli arteri telah dilaporkan berikutan suntikan perencat VEGF intravitreal.
Terdapat data terhad mengenai keselamatan rawatan DME, edema makula yang disebabkan oleh RVO dan CNV sekunder kepada PM pada pesakit dengan riwayat strok atau serangan iskemia sementara. Perhatian khusus harus diberikan semasa merawat pesakit tersebut (lihat bahagian 4.8).
Episod sebelumnya RVO, cabang iskemia dan RVO pusat
Terdapat pengalaman terhad dalam rawatan pesakit dengan episod RVO sebelumnya dan pesakit dengan RVO cabang iskemia (BRVO) dan RVO pusat (CRVO). Pada pesakit dengan RVO yang hadir dengan kehilangan fungsi visual dengan tanda-tanda klinikal iskemia tidak dapat dipulihkan, rawatan adalah tidak digalakkan.
04.5 Interaksi dengan produk ubat lain dan bentuk interaksi lain
Tidak ada kajian interaksi konvensional yang dilakukan.
Untuk gabungan penggunaan terapi fotodinamik (PDT) dengan verteporfin dan Lucentis pada AMD dan PM basah, lihat bahagian 5.1.
Untuk gabungan penggunaan photocoagulation laser dan Lucentis dalam rawatan DME dan BRVO, lihat bahagian 4.2 dan 5.1.
04.6 Kehamilan dan penyusuan
Wanita berpotensi melahirkan anak / kontrasepsi pada wanita
Wanita yang berpotensi melahirkan anak harus menggunakan alat kontrasepsi yang berkesan semasa rawatan.
Kehamilan
Untuk ranibizumab, tidak ada data klinikal mengenai kehamilan yang terdedah. Kajian pada monyet cynomolgus tidak menunjukkan kesan berbahaya langsung atau tidak langsung terhadap kehamilan atau perkembangan embrio / janin (lihat bahagian 5.3). Pendedahan sistemik kepada ranibizumab rendah berikutan pentadbiran okular, tetapi kerana mekanisme tindakan ranibizumab harus dianggap berpotensi teratogenik dan embrio / foetotoksik. Oleh itu, ranibizumab tidak boleh digunakan semasa mengandung kecuali manfaat yang diharapkan melebihi risiko yang mungkin timbul pada janin. Wanita yang merancang untuk hamil dan telah dirawat dengan ranibizumab disyorkan untuk menunggu sekurang-kurangnya 3 bulan setelah dos terakhir ranibizumab sebelum mengandung bayi.
Kehamilan
Tidak diketahui sama ada Lucentis diekskresikan dalam susu manusia. Sebaiknya jangan menyusu semasa menggunakan Lucentis.
Kesuburan
Tidak ada data mengenai kesuburan.
04.7 Kesan terhadap kemampuan memandu dan menggunakan mesin
Prosedur rawatan Lucentis boleh menyebabkan gangguan visual sementara yang boleh mempengaruhi kemampuan memandu atau menggunakan mesin (lihat bahagian 4.8). Pesakit yang mengalami gejala ini tidak boleh memandu atau mengoperasikan mesin sehingga gangguan visual sementara ini berhenti.
04.8 Kesan yang tidak diingini
Ringkasan profil keselamatan
Sebilangan besar reaksi buruk yang dilaporkan berikutan pemberian Lucentis berkaitan dengan prosedur suntikan intravitreal.
Reaksi buruk okular yang paling kerap dilaporkan selepas suntikan Lucentis adalah: sakit mata, hiperemia okular, peningkatan tekanan intraokular, vitreitis, detasmen vitreous, pendarahan retina, gangguan visual, floaters (vitreous floaters), pendarahan konjungtiva, mata iritasi, sensasi badan asing di mata, peningkatan koyakan, blepharitis, mata kering dan mata gatal.
Reaksi buruk bukan okular yang paling kerap dilaporkan adalah sakit kepala, nasofaringitis dan arthralgia.
Reaksi buruk yang kurang kerap dilaporkan tetapi lebih serius termasuk endophthalmitis, kebutaan, detasmen retina, pecah retina dan katarak trauma iatrogenik (lihat bahagian 4.4).
Pesakit harus dimaklumkan mengenai gejala-gejala reaksi buruk yang berpotensi ini dan diarahkan untuk memberitahu doktor mereka jika mereka mengalami tanda-tanda seperti sakit mata atau peningkatan ketidakselesaan, kemerahan mata yang semakin teruk, penglihatan kabur atau berkurang, peningkatan jumlah pelampung vitreous, atau " peningkatan kepekaan terhadap cahaya.
Reaksi buruk yang dilaporkan berikutan pemberian Lucentis dalam kajian klinikal diringkaskan dalam jadual di bawah.
Jadual tindak balas buruk #
Reaksi buruk disenaraikan mengikut kelas organ dan kekerapan menggunakan konvensyen berikut: sangat biasa (≥1 / 10), biasa (≥1 / 100,
Jangkitan dan jangkitan
Sangat biasa Nasofaringitis
biasa Jangkitan saluran kencing *
Gangguan sistem darah dan limfa
biasa Anemia
Gangguan sistem imun
biasa Hipersensitiviti
Gangguan psikiatri
biasa Keresahan
Gangguan sistem saraf
Sangat biasa Sakit kepala
Gangguan mata
Sangat biasa Vitreitis, detasmen vitreous, pendarahan retina, gangguan penglihatan, sakit mata, floaters vitreous, pendarahan konjungtiva, kerengsaan mata, sensasi badan asing di mata, peningkatan laserasi, blepharitis, mata kering, hiperemia okular, mata gatal.
biasa Degenerasi retina, gangguan retina, detasmen retina, robek retina, detasmen epitel pigmen retina, robekan epitel pigmen retina, ketajaman penglihatan berkurang, pendarahan vitreous, gangguan vitreous, uveitis, iritis, iridocyclitis, katarak, kapsul posterior katarak subkapsular, punct keratitis, lelasan kornea, reaksi ruang anterior, penglihatan kabur, pendarahan di tempat suntikan, pendarahan mata, konjungtivitis, konjungtivitis
alahan, pelepasan okular, kilatan bercahaya, fotofobia, ketidakselesaan okular, edema kelopak mata, sakit kelopak mata, hiperemia konjungtiva.
Tidak biasa Kebutaan, endophthalmitis, hypopion, hyphema, keratopathy, iris synechiae, deposit kornea, edema kornea, striae kornea, sakit di tempat suntikan, kerengsaan di tempat suntikan, sensasi yang tidak normal di mata, kerengsaan kelopak mata.
Gangguan pernafasan, toraks dan mediastinum
biasa Batuk
Gangguan saluran gastrousus
biasa Loya
Gangguan tisu kulit dan subkutan
biasa Reaksi alergi (ruam, gatal-gatal, gatal-gatal, eritema)
Gangguan tisu muskuloskeletal dan penghubung
Sangat biasa Arthralgia
Ujian diagnostik
Sangat biasa Tekanan intraokular meningkat
# Reaksi buruk didefinisikan sebagai kejadian buruk (sekurang-kurangnya 0.5 mata peratusan pesakit) yang berlaku pada kadar yang lebih tinggi (sekurang-kurangnya 2 mata peratusan) pada pesakit yang menerima rawatan dengan Lucentis 0,5 mg berbanding dengan mereka yang menerima rawatan kawalan (palsu atau PDT verteporfin).
* diperhatikan hanya pada populasi dengan DME
Reaksi buruk yang berkaitan dengan kategori ubat
Dalam kajian AMD basah fasa III, kekerapan keseluruhan pendarahan bukan okular, kejadian buruk yang berpotensi berkaitan dengan perencat VEGF (faktor pertumbuhan saluran endotel), sedikit meningkat pada pesakit yang dirawat dengan ranibizumab. Walau bagaimanapun, tidak ada. corak antara pendarahan yang berbeza. Terdapat risiko teoritis kejadian tromboemboli arteri, termasuk strok dan infark miokard, yang disebabkan oleh penggunaan perencat VEGF secara intravitreal. Kejadian rendah kejadian tromboemboli arteri diperhatikan dalam ujian klinikal dengan Lucentis pada pesakit dengan AMD, DME, RVO dan PM dan tidak ada perbezaan yang diperhatikan antara kumpulan ranibizumab berbanding dengan kawalan.
Melaporkan tindak balas buruk yang disyaki
Pelaporan tindak balas buruk yang disyaki yang berlaku setelah kebenaran produk ubat adalah penting, kerana memungkinkan pemantauan berterusan terhadap nisbah manfaat / risiko produk ubat tersebut.Para profesional kesihatan diminta melaporkan sebarang reaksi buruk yang disyaki melalui Badan Perubatan Itali. , laman web: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
04.9 Overdosis
Kes overdosis tidak sengaja telah dilaporkan dari ujian klinikal pada data AMD basah dan pasca pemasaran. Reaksi buruk yang paling sering dikaitkan dengan kes ini adalah peningkatan tekanan intraokular, kebutaan sementara, penurunan ketajaman penglihatan, edema kornea dan kesakitan. Sekiranya berlaku overdosis, tekanan intraokular harus dipantau dan dirawat sebagaimana yang difikirkan perlu oleh doktor.
05.0 HARTA FARMAKOLOGI
05.1 Sifat farmakodinamik
Kumpulan farmakoterapi: Oftalmologi, agen anti-neovaskular, kod ATC: S01LA04
Ranibizumab adalah fragmen antibodi monoklonal rekombinan manusia yang diarahkan terhadap faktor pertumbuhan endotel vaskular manusia A (VEGF-A). Ia mengikat dengan pertalian tinggi dengan isoform VEGF-A (contohnya VEGF110, VEGF121 dan VEGF165), sehingga menghalang pengikatan VEGF-A ke reseptor VEGFR-1 dan VEGFR-2nya. Reseptornya membawa kepada percambahan sel endotel neovaskularisasi, dan peningkatan kebolehtelapan vaskular, yang dianggap menyumbang kepada perkembangan bentuk neovaskular degenerasi makula yang berkaitan dengan usia, miopia patologi atau penurunan penglihatan yang disebabkan oleh edema makula diabetes atau "Edema makular sekunder kepada RVO.
Rawatan AMD basah
Untuk AMD basah, keselamatan dan keberkesanan klinikal Lucentis dinilai dalam tiga kajian terkawal secara rawak, double-blind, palsu atau aktif 24 bulan pada pesakit dengan AMD neovaskular. Sebanyak 1.323 pesakit (879 dirawat dan 444 kawalan) telah mendaftar dalam kajian ini.
Dalam kajian FVF2598g (MARINA), 716 pesakit dengan lesi neovaskularisasi choroidal klasik atau gaib (CNV) tanpa komponen klasik menerima suntikan intravitreal bulanan Lucentis 0.3 mg (n = 238) atau 0.5 mg (n = 240) atau suntikan palsu (n = 238).
Dalam kajian FVF2587g (ANCHOR), 423 pesakit dengan CNV terutamanya klasik menerima salah satu rawatan berikut: 1) suntikan intravitreal bulanan Lucentis 0.3 mg dan PDT palsu (n = 140); 2) suntikan intravitreal bulanan Lucentis 0.5 mg dan PDT palsu (n = 140); atau 3) suntikan palsu intravitreal dan PDT dengan verteporfin (n = 143). PDT dengan verteporfin atau sham diberikan bersama dengan suntikan awal Lucentis dan seterusnya setiap 3 bulan jika fluorangiografi menunjukkan kegigihan atau penyebaran semula kebocoran vaskular.
Penemuan utama diringkaskan dalam Jadual 1, 2 dan Gambar 1.
Jadual 1 Hasil pada bulan 12 dan bulan 24 dalam kajian FVF2598g (MARINA)
ap
Jadual 2 Hasil pada Bulan 12 dan Bulan 24 dalam Kajian FVF2587g (ANCHOR)
Jadual
Hasil kedua kajian menunjukkan bahawa rawatan lanjutan dengan ranibizumab juga bermanfaat bagi pesakit yang kehilangan ≥15 huruf ketajaman penglihatan (BCVA) terbaik pada tahun pertama rawatan.
Kajian FVF3192g (PIER) adalah kajian terkawal rawak, buta ganda, palsu yang dirancang untuk menilai keselamatan dan keberkesanan Lucentis pada 184 pesakit dengan semua bentuk AMD neovaskular. Pesakit menerima suntikan intravitreal Lucentis 0.3 mg (n = 60) atau 0.5 mg (n = 61) atau suntikan palsu (n = 63) sekali sebulan untuk 3 dos berturut-turut, diikuti dengan satu dos yang diberikan sekali setiap 3 bulan. Dari bulan 14 kajian, pesakit yang dirawat dengan suntikan palsu dimasukkan ke rawatan dengan ranibizumab dan dari bulan 19, rawatan yang lebih kerap dapat dilakukan. Pesakit yang dirawat dengan Lucentis dalam kajian PIER mendapat purata 10 rawatan secara keseluruhan.
Titik akhir keberkesanan utama adalah perubahan min dalam ketajaman penglihatan pada 12 bulan berbanding tahap awal. Setelah peningkatan awal dalam ketajaman penglihatan (mengikuti dos bulanan), rata-rata, ketajaman penglihatan pesakit menurun dengan dos suku tahun, kembali ke awal pada bulan 12 dan kesan ini dikekalkan pada kebanyakan pesakit yang dirawat. Dengan ranibizumab (82%) pada Bulan 24. Data dari sejumlah subjek yang telah dipindahkan ke rawatan ranibizumab setelah lebih dari satu tahun rawatan palsu menunjukkan bahawa permulaan rawatan awal mungkin dikaitkan dengan pengekalan ketajaman penglihatan yang lebih baik.
Dalam kedua-dua kajian MARINA dan ANCHOR, peningkatan ketajaman penglihatan yang diperhatikan dengan Lucentis 0,5 mg pada 12 bulan disertai dengan manfaat yang dilaporkan oleh pesakit yang diukur oleh skor National Eye Institute Visual Function Questionnaire (VFQ-25). Perbezaan antara Lucentis 0.5 mg dan dua kumpulan kawalan dinilai dengan nilai p antara 0,009 hingga
Keberkesanan Lucentis dalam rawatan AMD basah disahkan dalam kajian AMD pasca pemasaran. Data dari dua kajian (MONT BLANC, BPD952A2308 dan DENALI, BPD952A2309) tidak menunjukkan kesan tambahan. Daripada gabungan gabungan verteporfin (Visudyne PDT) dan Lucentis berbanding Lucentis sahaja.
Rawatan gangguan penglihatan akibat DME
Keselamatan dan keberkesanan Lucentis dinilai dalam dua kajian 12 bulan secara rawak, double-blind, terkawal palsu atau aktif pada pesakit dengan penglihatan yang menurun akibat edema makula diabetes. Sebanyak kajian ini didaftarkan. Daripada 496 pesakit (336 aktif dan 160 kawalan), kebanyakan menghidap diabetes jenis II, 28 pesakit yang dirawat mempunyai diabetes jenis I.
Pada fasa II kajian D2201 (RESOLVE), 151 pesakit dirawat dengan ranibizumab (6 mg / mL, n = 51, 10 mg / mL, n = 51) atau palsu (n = 49) dengan satu suntikan intravitreal setiap bulan. sehingga kriteria yang telah ditentukan tercapai.Dosis permulaan ranibizumab (0,3 mg atau 0,5 mg) dapat dua kali ganda pada bila-bila masa semasa kajian selepas suntikan pertama. Photocoagulation laser dibenarkan sebagai rawatan penyelamat dari bulan 3 di kedua-dua lengan rawatan. Kajian ini mempunyai dua bahagian: bahagian eksplorasi (42 pesakit pertama dikunjungi pada bulan 6) dan bahagian pengesahan (baki 109 pesakit yang dikunjungi pada bulan 12).
Penemuan utama dari bahagian pengesahan kajian (2/3 pesakit) diringkaskan dalam Jadual 3.
Jadual 3 Hasil pada Bulan 12 dalam Kajian D2201 (RESOLVE) (Jumlah Populasi Kajian)
ap
Dalam fasa III kajian D2301 (RESTORE), 345 pesakit dengan gangguan penglihatan akibat edema makula secara rawak menerima sama ada "suntikan intravitreal 0,5 mg ranibizumab sebagai monoterapi dan laser photocoagulation (n = 116), atau gabungan 0,5 mg ranibizumab dan laser photocoagulation (n = 118) atau "suntikan palsu dan laser photocoagulation (n = 111). Rawatan dengan ranibizumab dimulakan dengan suntikan intravitreal bulanan dan berlanjutan sehingga ketajaman penglihatan tetap stabil untuk sekurang-kurangnya tiga pemeriksaan bulanan berturut-turut. Rawatan disambung semula apabila penurunan BCVA akibat perkembangan DME diperhatikan. Fotocagulasi laser diberikan pada awal pada hari yang sama, sekurang-kurangnya 30 minit sebelum suntikan ranibizumab, dan selepas itu mengikut keperluan berdasarkan kriteria ETDRS.
Penemuan utama diringkaskan dalam Jadual 4 dan Gambar 2.
Jadual 4 Hasil pada Bulan 12 dalam Kajian D2301 (PULANGAN)
ap
Kesannya konsisten di kebanyakan subkumpulan. Walau bagaimanapun, subjek dengan BCVA yang cukup tinggi pada peringkat awal (> 73 huruf) dengan edema makula dan ketebalan retina pusat
Peningkatan ketajaman penglihatan pada Bulan 12 yang diperhatikan dengan Lucentis 0,5 mg disertai dengan manfaat pesakit yang dilaporkan mengenai fungsi berkaitan penglihatan utama yang diukur oleh skor National Eye Institute Visual Function Questionnaire (VFQ-25). Tidak ada perbezaan kerana rawatan dibentuk dalam subkelas soal selidik ini.Perbezaan antara Lucentis 0,5 mg dan kumpulan kawalan dinilai dengan nilai p 0,0137 (ranibizumab mono) dan 0,0041 (ranibizumab + laser) untuk skor komposit VFQ-25.
Dalam kedua-dua kajian, peningkatan visual disertai dengan penurunan berterusan edema makula yang diukur sebagai ketebalan retina pusat (CRT).
Rawatan gangguan penglihatan yang disebabkan oleh edema makula sekunder daripada RVO
Keselamatan dan keberkesanan klinikal Lucentis pada pesakit dengan gangguan penglihatan akibat edema makular sekunder RVO dinilai dalam percubaan rawak, double-blind, terkawal: BRAVO dan CRUISE yang merekrut pesakit dengan BRVO (n = 397) dan CRVO (n = 392 Dalam kedua-dua kajian, pesakit menerima suntikan 0,3 mg atau 0,5 mg ranibizumab intravitreal atau palsu. Selepas 6 bulan, pesakit di lengan kawalan palsu dipindahkan ke kumpulan ranibizumab 0,5 mg. Dalam kajian BRAVO, laser photocoagulation sebagai rawatan penyelamat dibenarkan di semua pelukan dari bulan 3.
Penemuan utama dari kajian BRAVO dan CRUISE ditunjukkan dalam Jadual 5 dan 6
Jadual 5 Hasil pada bulan 6 dan 12 (BRAVO)
ap
Jadual 6 Hasil pada Bulan 6 dan 12 (CRUISE)
ap
Dalam kedua-dua kajian, peningkatan visual disertai dengan pengurangan edema makula yang berterusan dan signifikan yang diukur dari segi ketebalan retina pusat.
Pada pesakit BRVO (kajian BRAVO dan pelanjutan kajian HORIZON): Selepas 2 tahun, pesakit yang telah dirawat dengan suntikan palsu dalam 6 bulan pertama dan kemudian dipindahkan ke rawatan ranibizumab mempunyai peningkatan AV (& simp; 15 huruf) setanding dengan itu pesakit yang telah dirawat dengan ranibizumab sejak permulaan kajian (& simp; 16 huruf). Walau bagaimanapun, jumlah pesakit yang menyelesaikan 2 tahun adalah terhad dan hanya lawatan suku tahun yang dijadualkan dalam kajian HORIZON. ada bukti yang mencukupi untuk disimpulkan dengan cadangan mengenai apabila rawatan ranibizumab harus dimulakan pada pesakit dengan BRVO.
Pada pesakit CRVO (kajian CRUISE dan pelanjutan kajian HORIZON): Selepas 2 tahun, pesakit yang telah dirawat pada 6 bulan pertama dengan suntikan palsu dan kemudian dipindahkan ke rawatan ranibizumab tidak menunjukkan peningkatan AV (& simp; 6 huruf) berbanding dengan pesakit yang telah dirawat dengan ranibizumab sejak permulaan kajian (& simp; 12 huruf).
Peningkatan ketajaman penglihatan yang diperhatikan dengan rawatan ranibizumab pada bulan 6 dan 12 disertai dengan manfaat yang dilaporkan oleh pesakit yang diukur oleh subkumpulan National Eye Institute Visual Function Questionnaire (NEI VFQ-25) aktiviti dekat dan jauh Perbezaan antara Lucentis 0.5 mg dan kumpulan kawalan didapati berada di antara nilai p antara 0.02 hingga 0.0002.
Rawatan gangguan penglihatan kerana CNV sekunder hingga PM
Keselamatan dan keberkesanan klinikal Lucentis pada pesakit dengan gangguan penglihatan akibat CNV pada PM disahkan berdasarkan data 12 bulan dari kajian rawak, buta ganda, terkawal F2301 (RADIANCE). Kajian ini bertujuan untuk menilai Dua rejimen dos yang berbeza ranibizumab 0,5 mg diberikan dengan suntikan intravitreal berbanding verteporfin PDT (vPDT, terapi fotodinamik Visudyne). 277 pesakit secara rawak menggunakan salah satu lengan berikut:
• Kumpulan I (ranibizumab 0,5 mg, rejimen rawatan ditentukan oleh kriteria "kestabilan" yang ditakrifkan sebagai tidak ada perubahan dalam BCVA dibandingkan dengan penilaian dua bulan sebelumnya).
• Kumpulan II (ranibizumab 0,5 mg, rejimen rawatan ditentukan oleh kriteria "aktiviti penyakit" yang ditakrifkan sebagai gangguan penglihatan yang disebabkan oleh cairan intra- atau subretinal atau kebocoran aktif yang disebabkan oleh lesi CNV seperti yang dibuktikan oleh OCT dan / atau AF).
• Kumpulan III (pesakit yang dirawat dengan vPDT - dengan kemungkinan rawatan dengan ranibizumab mulai bulan 3).
Selama 12 bulan kajian, pesakit menerima rata-rata 4.6 suntikan (julat 1-11) pada Kumpulan I dan 3.5 suntikan (julat 1-12) pada Kumpulan II. Di antara pesakit yang tergolong dalam Kumpulan II, yang menunjukkan posologi yang disarankan (lihat bahagian 4.2), 50.9% pesakit menjalani rawatan dengan 1 hingga 2 suntikan, 34.5% 3 hingga 5 suntikan dan 14.7% memberikan 6 hingga 12 suntikan selama 12 bulan kajian . 62.9% pesakit Kumpulan II tidak memerlukan suntikan selama 6 bulan kedua kajian.
Penemuan utama dari RADIANCE diringkaskan dalam Jadual 7 dan Gambar 5.
Jadual 7 Hasil pada Bulan 3 dan 12 (RADIANCE)
ap
b Pengendalian perbandingan hingga bulan 3. Pesakit secara rawak untuk menerima vPDT layak mendapat rawatan ranibizumab pada bulan 3 (dalam Kumpulan III, 38 pesakit menerima ranibizumab pada bulan 3)
Peningkatan penglihatan disertai dengan pengurangan ketebalan retina pusat.
Berbanding dengan kumpulan yang dirawat vPDT, pesakit dalam kumpulan yang dirawat ranibizumab melaporkan manfaat (nilai p
Populasi kanak-kanak
Keselamatan dan keberkesanan ranibizumab pada kanak-kanak belum dapat dipastikan.
Agensi Ubat-ubatan Eropah telah mengetepikan kewajiban untuk menyerahkan hasil kajian dengan Lucentis di semua subset populasi pediatrik untuk AMD neovaskular, gangguan penglihatan akibat DME, gangguan penglihatan akibat edema makular sekunder kepada RVO dan gangguan penglihatan kerana CNV sekunder hingga PM (lihat bahagian 4.2 untuk maklumat mengenai penggunaan pediatrik).
05.2 Sifat farmakokinetik
Selepas pemberian Lucentis intravitreal bulanan kepada pesakit dengan AMD neovaskular, kepekatan serum ranibizumab pada amnya rendah, dengan tahap maksimum (Cmax) umumnya di bawah kepekatan ranibizumab yang diperlukan untuk menghalang aktiviti biologi VEGF sebanyak 50% (11-27 ng / mL , dinilai dalam ujian secara in vitro percambahan sel). Cmax adalah berkadar dosis sepanjang julat dos antara 0,05 hingga 1,0 mg / mata. Dalam jumlah pesakit DME yang terhad, kepekatan serum yang dikesan menunjukkan bahawa pendedahan sistemik yang sedikit lebih tinggi tidak dapat dikesampingkan. Kepekatan serum ranibizumab pada pesakit dengan RVO adalah serupa atau sedikit lebih tinggi daripada yang diamati pada pesakit dengan AMD neovaskular.
Berdasarkan analisis farmakokinetik populasi dan pembersihan serum ranibizumab untuk pesakit AMD neovaskular yang dirawat dengan dos 0.5 mg, min jangka hayat penghapusan vitreous ranibizumab adalah kira-kira 9 hari. Pada masa pentadbiran intravitreal bulanan Lucentis 0,5 mg / mata, serum C ranibizumab, mencapai kira-kira 1 hari selepas dos, secara amnya berkisar antara 0,79 dan 2,90 ng / ml, sementara Cmin pada umumnya berfluktuasi antara 0,07 dan 0,49 ng / ml. Kepekatan serum ranibizumab dianggarkan sekitar 90,000 kali lebih rendah daripada kepekatan vitreous.
Pesakit dengan kekurangan buah pinggang: Tidak ada kajian konvensional yang dilakukan untuk memeriksa farmakokinetik Lucentis pada pesakit dengan kekurangan buah pinggang. Dalam "analisis farmakokinetik pada populasi pesakit AMD neovaskular, 68% (136 daripada 200) pesakit mengalami" kekurangan buah pinggang (46.5% ringan [50-80 mL / min], 20% sederhana [30 -50 mL / min] dan 15% teruk [pelepasan sistemik sedikit lebih rendah, tetapi ini tidak signifikan secara klinikal.
Pesakit dengan kekurangan hati: Tidak ada kajian konvensional yang dilakukan untuk memeriksa farmakokinetik Lucentis pada pesakit dengan kekurangan hati.
05.3 Data keselamatan praklinikal
Pemberian ranibizumab intravitreal dua hala kepada monyet cynomolgus pada dos antara 0.25 mg / mata dan 2.0 mg / mata sekali setiap 2 minggu sehingga 26 minggu mengakibatkan kesan okular bergantung kepada dos.
Secara intraokular, peningkatan suar dan sel yang bergantung pada dos berlaku di ruang anterior, memuncak 2 hari selepas suntikan. Keterukan tindak balas keradangan umumnya menurun dengan suntikan berikutnya atau semasa tempoh pemulihan. Pada segmen posterior Penyusupan selular dan floaters vitreous berlaku, yang juga cenderung bergantung pada dos dan umumnya berterusan sehingga akhir tempoh rawatan.Dalam kajian 26 minggu, keparahan keradangan vitreous meningkat dengan jumlah suntikan. Walau bagaimanapun, kebolehbalikan diperhatikan selepas tempoh pemulihan. Sifat dan jangka masa keradangan segmen posterior menunjukkan tindak balas antibodi yang dimediasi oleh imun, yang mungkin tidak relevan secara klinikal. Pembentukan katarak telah diamati pada beberapa haiwan setelah peradangan intensif yang cukup lama, menunjukkan bahawa perubahan lensa adalah sekunder kepada keradangan yang teruk. Peningkatan sementara tekanan intraokular diperhatikan selepas pentadbiran, tanpa mengira dos, selepas suntikan intravitreal.
Perubahan okular mikroskopik berkaitan dengan keradangan dan tidak menunjukkan proses degeneratif. Perubahan granulomatosa keradangan dicatat pada cakera optik beberapa mata. Perubahan segmen posterior ini berkurang, dan dalam beberapa kes diselesaikan, selama tempoh pemulihan.
Tidak ada tanda-tanda ketoksikan sistemik berikutan pentadbiran intravitreal. Antibodi serum dan vitreous terhadap ranibizumab dijumpai pada subset haiwan yang dirawat.
Tidak ada data karsinogenisiti atau mutagenisiti.
Pada monyet hamil, suntikan ranibizumab secara intravitreal mengakibatkan pendedahan sistemik maksimum 0.9-7 kali pendedahan klinikal terburuk tidak menyebabkan ketoksikan perkembangan atau teratogenik, dan tidak memberi kesan pada berat badan atau struktur badan. Plasenta, walaupun ranibizumab harus dianggap berpotensi teratogenik dan embrio / foetotoksik berdasarkan kesan farmakologinya.
Ketiadaan kesan ranibizumab yang dimediasi pada perkembangan janin / janin banyak berkaitan terutamanya dengan ketidakmampuan fragmen Fab untuk menyeberangi plasenta. Walau bagaimanapun, satu kes dijelaskan dengan tahap ranibizumab serum ibu yang tinggi dan kehadiran ranibizumab dalam serum janin, menunjukkan bahawa antibodi anti-ranibizumab bertindak sebagai protein (yang mengandungi wilayah FC) yang mengangkut ranibizumab, sehingga menurunkan penghapusannya dari serum ibu dan membenarkan pemindahannya ke plasenta. Oleh kerana ujian terhadap perkembangan embrio / janin telah dilakukan pada haiwan hamil yang sihat dan beberapa penyakit (seperti diabetes) dapat mengubah kebolehtelapan plasenta menjadi fragmen Fab, kajian itu mesti ditafsirkan dengan berhati-hati.
06.0 MAKLUMAT FARMASI
06.1 Eksipien
α, α-trehalosa dihidrat
Histidine hidroklorida, monohidrat
Histidin
Polysorbate 20
Air untuk suntikan
06.2 Ketidaksesuaian
Sekiranya tidak ada kajian keserasian, produk ubat ini tidak boleh dicampurkan dengan produk ubat lain.
06.3 Tempoh sah
3 tahun
06.4 Langkah berjaga-jaga khas untuk penyimpanan
Simpan di dalam peti sejuk (2 ° C - 8 ° C).
Jangan beku.
Simpan botol dalam kadbod luar untuk melindungi ubat daripada cahaya.
06.5 Sifat pembungkusan segera dan kandungan bungkusan
Larutan steril 0.23 ml dalam botol (kaca jenis I) dengan penyumbat (getah klorobutil), 1 jarum penapis tumpul (18G x 1½ ", 1.2 mm x 40 mm, 5 mcm), 1 jarum suntikan (30G x ½", 0.3 mm x 13 mm) dan 1 picagari (polipropilena) (1 ml). Pek mengandungi 1 botol.
06.6 Arahan penggunaan dan pengendalian
Botol, jarum suntikan, jarum penapis dan jarum suntik hanya untuk penggunaan tunggal. Penggunaan semula boleh menyebabkan jangkitan atau penyakit / kecederaan lain. Semua komponen steril. Sebarang komponen dengan kemasan yang menunjukkan tanda kerosakan atau gangguan tidak boleh digunakan. Kemandulan tidak dapat dijamin jika meterai pembungkusan komponen tidak utuh.
Untuk menyediakan Lucentis untuk suntikan intravitreal, ikuti arahan di bawah:
1. Keluarkan bahagian luar penyumbat getah botol sebelum dikumpulkan.
2. Pasangkan jarum penapis 5 mcm secara aseptik (18G x 1½ ", 1.2 mm x 40 mm, dibekalkan) ke picagari 1 ml (disediakan). Masukkan jarum penapis tumpul ke bahagian tengah penutup sehingga menyentuh bahagian bawah botol.
3. Tarik semua cecair dari botol dengan menahannya dalam kedudukan tegak, sedikit miring untuk memudahkan pengeluaran sepenuhnya.
4. Pastikan pelocok jarum suntikan ditarik ke belakang cukup jauh semasa mengosongkan botol untuk mengosongkan sepenuhnya jarum penapis.
5. Biarkan jarum penapis tumpul di dalam botol dan keluarkan jarum suntik darinya.Buang jarum penapis setelah mengeluarkan isi botol dan jangan gunakannya untuk suntikan intravitreal.
6. Pasangkan jarum suntikan (30G x ½ ", 0.3mm x 13mm, dibekalkan) dengan selamat dan aseptik ke picagari.
7. Tanggalkan penutup dari jarum suntikan dengan teliti tanpa melepaskan jarum suntikan dari jarum suntikan.
Catatan: Pegang asas jarum suntikan berwarna kuning sambil melepaskan penutupnya.
8. Keluarkan udara dengan hati-hati dari jarum suntik dan sesuaikan dos hingga 0,05 ml yang ditandakan pada jarum suntik.Suntik siap untuk disuntik.
Catatan: Jangan bersihkan jarum suntikan. Jangan tarik kembali pelocok.
Selepas suntikan, jangan tutup jarum atau lepaskan dari jarum suntik. Buangkan jarum suntik yang digunakan bersama jarum dalam bekas yang sesuai atau sesuai dengan keperluan tempatan.
07.0 PEMEGANG KEBENARAN PEMASARAN
Novartis Europharm Limited
Jalan Wimblehurst
Horsham
Sussex Barat, RH12 5AB
UK
08.0 NOMBOR KEBENARAN PEMASARAN
EU / 1/06/374/001
037608027
09.0 TARIKH KEBENARAN ATAU PEMBAHARUAN KEBENARAN
Tarikh kebenaran pertama: 22 Januari 2007
Tarikh pembaharuan terkini: 24 Januari 2012
10.0 TARIKH SEMAKAN TEKS
05/2014